
| High-Pressure Studies of Noncovalent Interactions in Supramolecular Atchitectures |
| From: PublishDate:2013-06-15 Hits: |
Supramolecular chemistry has attracted considerable attention since the conception was proposed. It is the interdiscipline subject that concerns with synthesis, properties and applications of “various molecular assemblies”. The supramolecular interactions, including hydrogen bonding and π‒stacking, play a critical role in crystal engineering. Noncovalent interactions display higher compressibility than covalent bonds. High pressure studies of noncovalent interactions and their cooperativity in the stability of supramolecular architectures are of fundamental and practical importance. To our knowledge, there are comparatively few investigations on mechanical properties of supramolecular architectures. Research on noncovalent interactions and their cooperativity in supramolecular materials under high-pressure conditions can make a contribution to understanding the structural resilience and robustness of supramolecular systems, which are crucial to optimal properties of the envisaged practical applications. We have undertaken a lot of works on structural properties of supramolecular architectures under high pressure. (1) Guanidinium Methanesulfonate (C(NH2)3+·CH3SO3−, GMS) Supramolecular architectures based on guanidinium cation and sulfonate anions RSO3− occupy an important position in the supramolecular chemistry area. Hundreds of layered guanidinium motifs have been synthesized on the basis of monosulfonate and disulfonate anions. The integrity of hydrogen-bonded sheet is attributed to the maximization of hydrogen bonds between six electron lone pairs on sulfonate oxygen atoms and six guanidinium protons, as well as electrostatic interactions between oppositely charged ions. Herein we choose guanidinium methanesulfonate as an ideal model to investigate the structural properties of guanidinium sulfonate motifs owing to its simpleness. We report the high-pressure response of guanidinium methanesulfonate using in situ Raman spectroscopy and synchrotron X-ray diffraction (XRD) techniques up to the pressures of ~11 GPa. GMS exhibits the representative supramolecular structure of two-dimensional (2D) hydrogen-bonded bilayered motifs under ambient conditions (Figure 1).
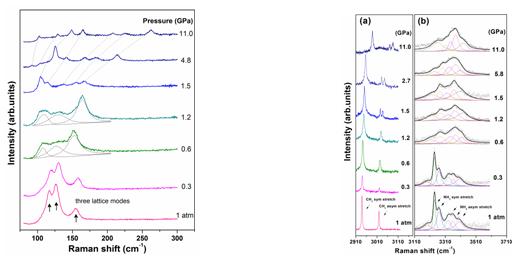
Figure 1 Crystal structure of GMS under ambient conditions. Figure 2 summarizes the evolution of the lattices modes of GMS. The three modes display ordinary blue shifts up to 0.6 GPa, originating from the expected contraction of interionic distances with increasing pressure. At 0.6 GPa, a new set of lattice modes emerges, indicating that GMS undergoes a phase transition from phase I to phase II. At 1.5 GPa, another set of lattice modes is observed, indicative of the second transition from phase II to phase III. The evolution of the lattice modes in phase III does not reveal any remarkable changes from 1.5 to 11.0 GPa, suggesting the phase III is stable up to 11.0 GPa. Typical Raman spectra in the range of 2910–3120 cm-1 and 3110–3600 cm-1 are shown in Figure 3. The two CH3 stretching modes reveal blue shifts owing to the repulsive nature of chemical environment around methyl groups below 0.6 GPa. The NH stretching modes exhibit the overall red shifts in the range of 0–0.6 GPa, implying the hydrogen bonds in phase I are of weak or moderate type. At 0.6 GPa, there is a pronounced redistribution in position and intensity in the NH stretching region, suggesting that the connectivity of hydrogen-bonded networks of guanidinium cations and methanesulfonate anions has been changed. Hence, the first phase transition may involve reconstruction of the two building blocks. At 1.5 GPa, the splitting of CH3 asymmetric stretching mode was traced, while the NH stretching modes do not involve any significant changes. Furthermore, the NH stretching patterns above 1.5 GPa are identical to those in phase II. These results demonstrate the second phase transition is not reconstructive and related to the distortions of methyl groups.
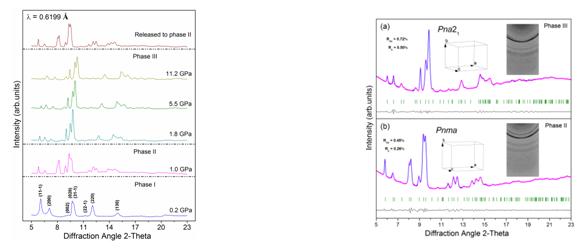
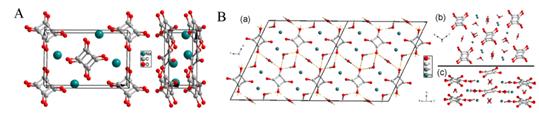
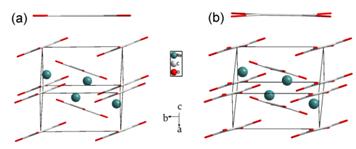
Figure 2 Selected Raman spectra of GMS lattice modes Figure 3 Selected Raman spectra of GMS in: (a) the range of 2910–3120 cm-1; and (b) the NH stretching region. To gain straightforward evidence and more information about the phase transitions, we performed synchrotron ADXRD experiments. Figure 4 presents the dependence of typical diffraction patterns of GMS as a function of pressure up to 11.2 GPa. As can be seen, the pattern at 1.0 GPa is quite distinct from the one of the ambient phase, signifying the transition from phase I to phase II. The diffraction pattern for the retrieved sample can be well indexed with Pnma symmetry, and the refined lattice parameters are a = 10.65(6) Å, b = 7.94(8) Å, c = 7.44(5) Å and V = 630.51(4) Å3 (Figure 5). Therefore, we propose the space group of phase II is Pnma. With further compression, the new set of diffraction pattern at 1.8 GPa implies the phase II of GMS has gone through a transition to phase III. No distinct change in the diffraction patterns is observed with further compression, proving the phase III to be stable up to the pressure of 11.2 GPa. The Pawley refinement of the pattern at 2.7 GPa reveals that the space group Pna21 matches the diffraction data very well. Hence, the phase III is proposed to possess Pna21 symmetry, and the indexed parameters are a = 9.70(5) Å, b = 7.42(5) Å, c = 7.82(4) Å and V = 563.82(7) Å3. Additionally, GMS is quenched to the phase II upon complete decompression, implying the first phase transition is irreversible and the second one is reversible. Figure 4 Representative synchrotron X-ray diffraction patterns of GMS Figure 5 Pawley refinements of GMS: (a) at 2.7 GPa; and (b) the pattern after releasing the pressure. (2) Sodium Squarate Under high pressure, the influence of π–stacking is always overwhelmed or affected by other interactions. And among the three kinds of stacking interaction, face-to-face π–stacking is the rarest one, due to its repulsive nature. These two points lead to the fact that the high-pressure exploration of face-to-face π–stacking is far from complete. Sodium Squarate (Na2C4O4, SS) possesses a rare but typical structure, based on face-to-face π–stacking interaction. It exhibits the unique structure with layers of squarate anions and sodium cations (Figure 1A). Face-to-faceπ–stacking interaction acts as repulsive force between the adjacent squarate planes. Meanwhile, electrostatic interactions also exist between the oppositely charged anions and cations to balance π–stacking. Squarate trihydrate (Na2C4O4•3H2O, SST) can be regarded as a deformation of SS, which is caused by the existence of water. Water complicates the layered structure of SS, but the fundamental face-to-face π–stacking structure has been retained (Figure 1B). We carry out in situ high-pressure Raman scattering and angle-dispersive synchrotron X-ray diffraction (ADXRD) studies of SS and SST, so as to detect the unique high-pressure behaviors of π–stacking, show how hydrogen bonding affects the behaviors of π–stacking under high pressure and explore the stability this series of stacked structures.
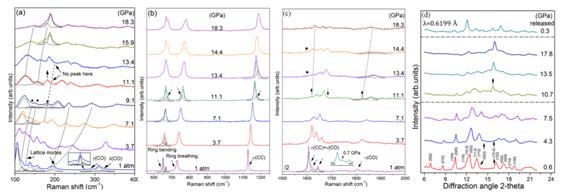
From the representative Raman and XRD patterns (Figure 2), we conducted that SS undergoes a phase transition at ~11.1 GPa, identified as a symmetry transformation from P21/c to P21. The complex behaviors of the ring modes in Raman spectra indicate that squarate rings are distorted by the increasing pressure. Perpendicular to the adjacent layers of squarate rings, it is the π–π repulsion that exists between them. Compression gives rise to the pacing reduction between the adjacent squarate planes and approach and overlap of the π-electron clouds over the squarate rings.The closer π-electron clouds result in the larger π–π repulsion between adjacent rings. When the strengthening of π–stacking cannot afford the increased free energy anymore, SS begins to adopt a new conformation. Therefore, we concluded that the enhancement of π–stacking between adjacent layers leads to the distortion of squarate rings, which is responsible for the phase transition at 11.1 GPa.
Figure 2(a)-(c) Representative high-pressure Raman spectra of SS. (d) Representative XRD patterns of SS at high pressures. In order to understand the mechanism of the phase transition more clearly, ab initio calculations were performed with the pseudopotential plane-wave method based on density functional theory. This calculation mode is coincident with our conclusion, deduced from the observed Raman spectra. As can be seen, the fundamental structure of SS still remains at high pressure. However, squarate rings are distorted by the increasing pressure, which results into the complex behaviors of the ring modes in Raman spectra. Meanwhile, in the calculated structural model, the decreasing of π–stacking related parameters indicates the enhancement of π–stacking interactions, which are consistent with the inferred mechanism of the phase transitions.
Figure 3a. Crystal structure of SS under ambient conditions. 3b Calculated structure of SS at 15 GPa. For comparison, high-pressure investigations on SST are performed up to 17.1 GPa. SST undergoes a phase transition at 10.3 GPa. As we have analyzed in SS, the squarate rings in SST are also be distorted by the increasing pressure. Due to the similar variations in Raman patterns and the analogical structures of SS and SST, we infer this distortion can also be caused by the enhancement of π–stacking. Whereas, with the effects of the strengthen hydrogen bonds, the distortions of the squarate rings in SST are not as obvious as in SS. Moreover, after the phase transition, the distorted squarate rings should be surrounded by the disordered water molecules. Our conclusion is also supported by high-pressure behavior of ammonium squarate ((NH4)2C4O4, AS). Comparing the high-pressure behaviors of AS, SS and SST, it can be inferred that the rearranged hydrogen bonding in AS complicates the distortion of squarate rings, but the only strengthened hydrogen bonds in SST restrain this change. Furthermore,hydrogen bonding can drag the ammonium ions and squarate anions in AS to form the disordered structure. However, there is no effect of hydrogen bonding in SS, so SS can keep in order after the phase transition. This is also the reason for the appearance of the disordered water in SST under high pressure. Additionally, the phase transitions of AS and SS are all reversible. So it is the electrostatic interaction that compels the compounds returning to their original structure. Furthermore, the structures of SS, SST, and AS are stabilized with face-to-face π–stacking. We conclude that the ambient phase of this series of materials can be stable up to ∼11 GPa.
Article: 1. Shourui Li, Qian Li, Jing Zhou, Run Wang, Zhangmei Jiang, Kai Wang, Dapeng Xu, Jing Liu, Bingbing Liu, Guangtian Zou, and Bo Zou,* Effect of High Pressure on the Typical Supramolecular Structure of Guanidinium Methanesulfonate, The Journal of Physical Chemistry B, 2012, 116(10), 3092-3098 2. Qian Li, Shourui Li, Kai Wang, Wenbo Li, Jing Liu, Bingbing Liu, Guangtian Zou, and Bo Zou,* Compression Studies of Face-to-Face π-Stacking Interaction in Sodium Squarate salts: Na2C4O4 and Na2C4O4.3H2O, The Journal of Chemical Physics, 2012, 137(18), 184905 |
|
|
| Chinese
Science Highlights
Home /


Copyright © 2011 - 2012 Beijing Synchrotron Radiation Facility